Click on an image to view larger version & data in a new window
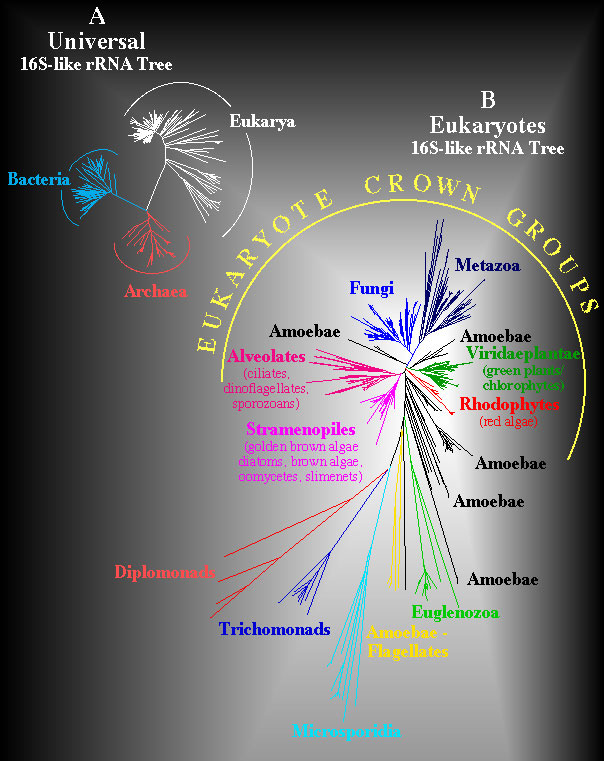
Universal and Eukaryote Phylogenetic Trees. Structural similarites for 900 sites that can be unambiguously aligned in a data set of more than 800 eukaryotes and 100 prokaryotes were computed and converted to evolutionary distances using the Kimura two parameter model. The Neighbor-Joining method was used to infer the unrooted universal phylogeny shown in A. A similar computation was used to infer an unrooted phylogeny in B for diverse eukaryotes but this analysis is based upon comparisons of 1200 positions that can be unambiguously aligned. Evolutionary distances in these trees are proportional to length of line segments separating taxa.